GxP Dictionary
A
A0-Value
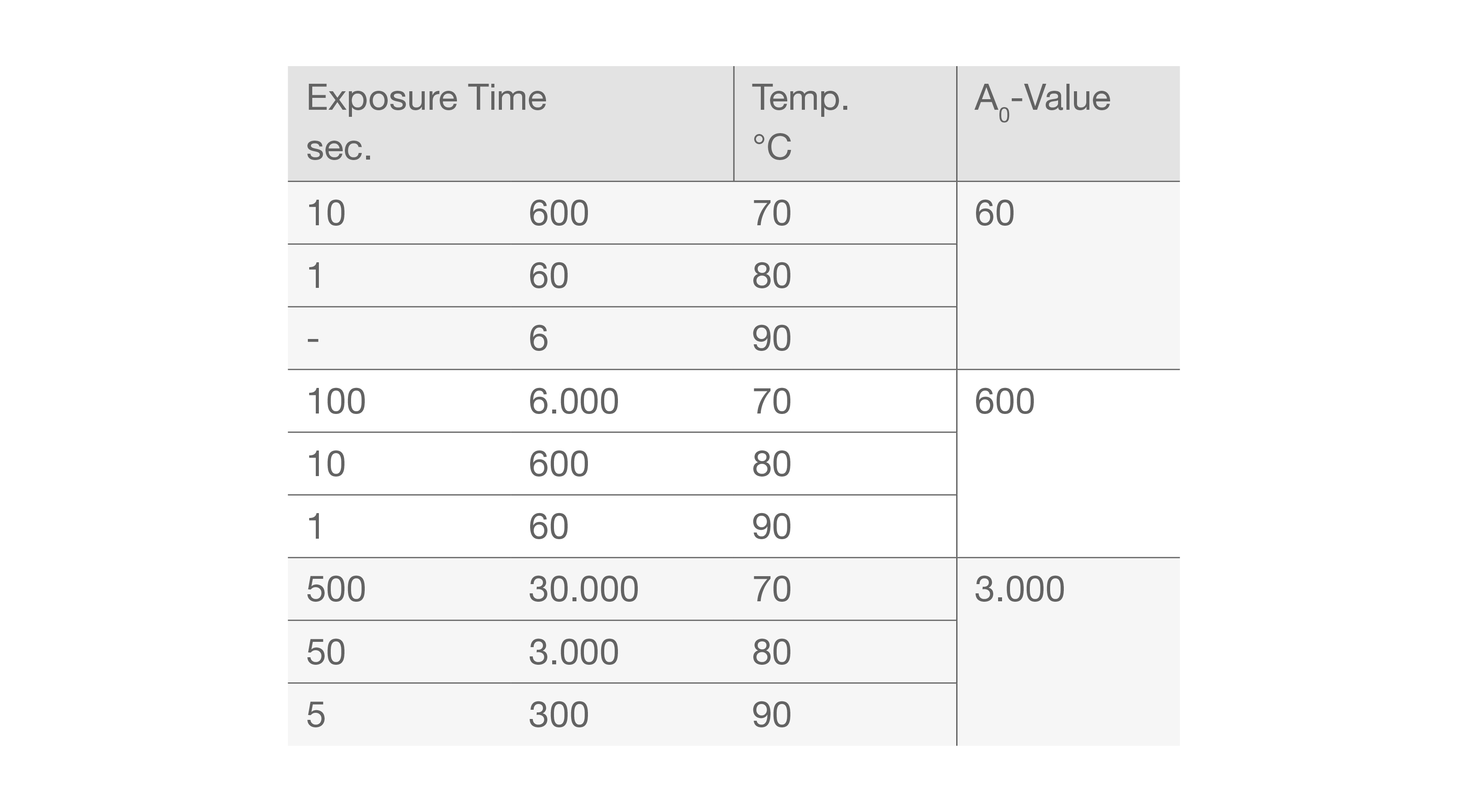
A
ALCOA (++) Principle
A
Annex 1
A
API – Active Pharmaceutical Ingredient
A
APR – Annual Product Review
A
Audit
A
Audit Trail
B
Bioburden
B
BP – British Pharmacopoeia
C
21 CFR – Code of Federal Regulations
C
21 CFR part11
C
Calibration
C
Calibration Protocol
C
CAPA – Corrective and Preventive Action
C
CC – Change Control
C
CFR – Code Federal Regulations
C
CFU – Colony Forming Unit
C
ChP – Chinese Pharmacopoeia
C
CIP – Clean in Place
C
Cold Chain
C
Cold Chain Management
C
Compliance
C
Concurrent Validation
C
CPV – Continued Process Validation
C
CQA – Critical Quality Attributes
C
CSV – Computer-System/ -Software-Validation
D
D-Value
D
Data Integrity
D
Design Specifications
D
Deviation
D
Deviation Management
D
DMS – Document Management System
D
DQ – Design Qualification
E
EMA – European Medicines Agency
E
EP – or Ph.Eur European Pharmacopeia
E
EU-GMP – Annex
E
EU-GMP Annex 11 – Computerized Systems
E
EU-GMP Annex 15 – Qualification and Validation
E
EU-GMP Annex 20 – Quality Risk Management (QRM)
E
EU-GMP Guidelines
F
F0-Value
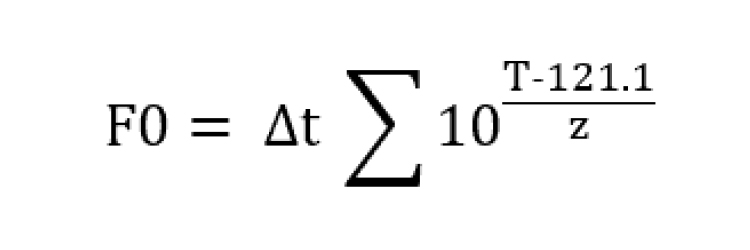
F
FAT – Factory-Acceptance-Test
F
FDA – 482
F
FDA – 483
F
FDA – Food and Drug Administration
F
FDA – Warning Letter
F
FDA Guidance for Industry – Process Validation
F
Fh-Value
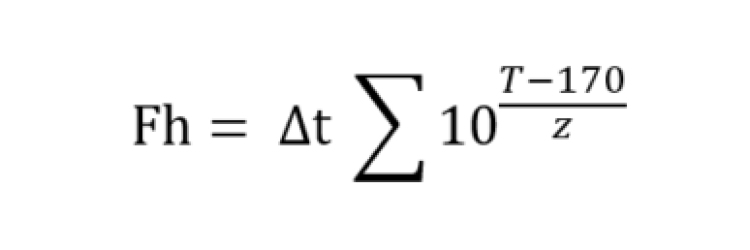
F
FMEA – Failure Mode and Effects Analysis
F
FMECA – Failure Mode, Effects, and Criticality Analysis
F
FTA – Fault Tree Analysis
G
GAMP® – Good Automated Manufacturing Practice
G
GAMP® 5V-Model

G
GCP – Good Clinical Practice
G
GDP – Good Distribution Practice
G
GEP – Good Engineering Practice
G
GLP – Good Laboratory Practice
G
GMP – Good Manufacturing Practice / cGMP – current Good Manufacturing Practice
G
GSP – Good Storage Practices
G
GxP/ cGxP
H
HACCP – Hazard Analysis and Critical Control Points
H
HAZOP – Hazard and Operability Study
I
ICH – International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use
I
ICH Q9 – Quality Risk Management – Scientific Guideline
I
IQ – Installation Qualification
I
ISPE® – International Society for Pharmaceutical Engineering
I
ISPE® GAMP® 5 – A Risk-Based Approach to Compliant GxP Computerized Systems
J
JP – Japanese Pharmacopeia
L
LVP – Large Volume Parenteral
O
OOE – Out-of-Expectation
O
OOS – Out-of-Specification
O
OOT – Out-of-Trend
O
OQ – Operational Qualification
P
PAT – Process Analytical Technology
P
PCS – Process Control System
P
PDA® Parenteral Drug Association
P
PHA – Preliminary Hazard Analysis
P
PIC/S® – Pharmaceutical Inspection Cooperation Scheme
P
PPQ – Process Performance Qualification
P
PQ – Performance Qualification
P
PQR – Product Quality Review
P
Predictive Maintenance
P
Preventive Maintenance
P
Prospective Validation or Premarket Validation
P
PS – Purified Steam
P
PV – Process Validation
P
PW – Purified Water
Q
QA – Quality Assurance
Q
QbD – Quality-by-Design
Q
QC – Quality Control
Q
QMS – Quality Management System
Q
QP – Qualification Protocol
Q
QP – Qualified Person
Q
QSM – Quality System Manual
Q
Qualification
R
Retrospective Validation
R
Revalidation
R
Risk Acceptance
R
Risk Analysis
R
Risk Assessment
R
Risk Communication
R
Risk Control
R
Risk Evaluation
R
Risk Identification
R
Risk Management
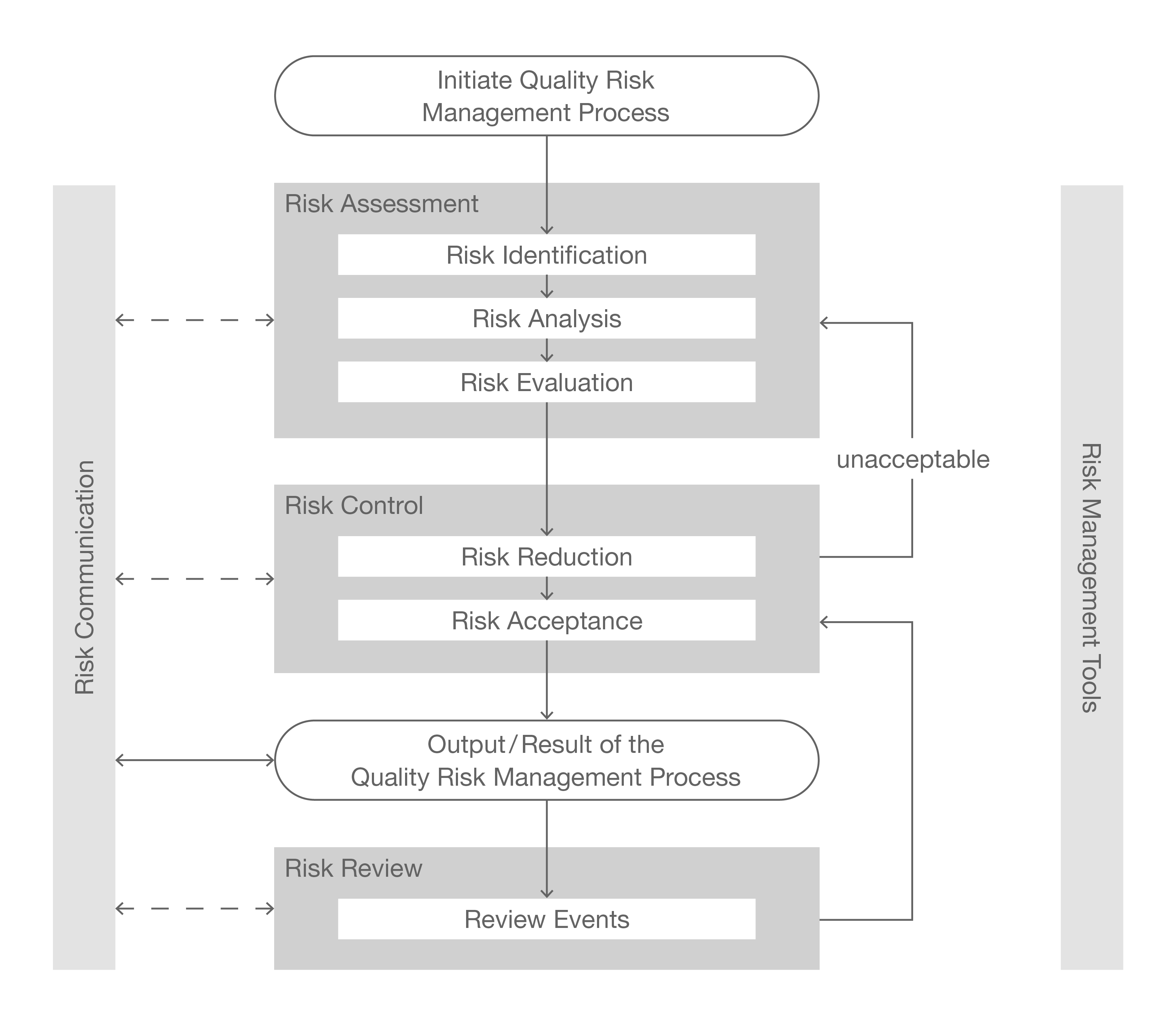
R
Risk Management Tools
R
Risk Reduction
R
Risk Review
S
SAL – Sterility Assurance Level
S
Sanitization
S
SAT – Site-Acceptance-Test
S
SIP – Steam in Place
S
SMF – Site Master File
S
SOP – Standard Operation Procedure
S
Steam Quality
S
SVPs – Small Volume Parenteral
T
Temperature (Heat) Penetration Study
T
Temperature Mapping Study
T
Traceability
U
URS – User-Requirements-Specifications
U
USP® – United States Pharmacopeia
V
Validation
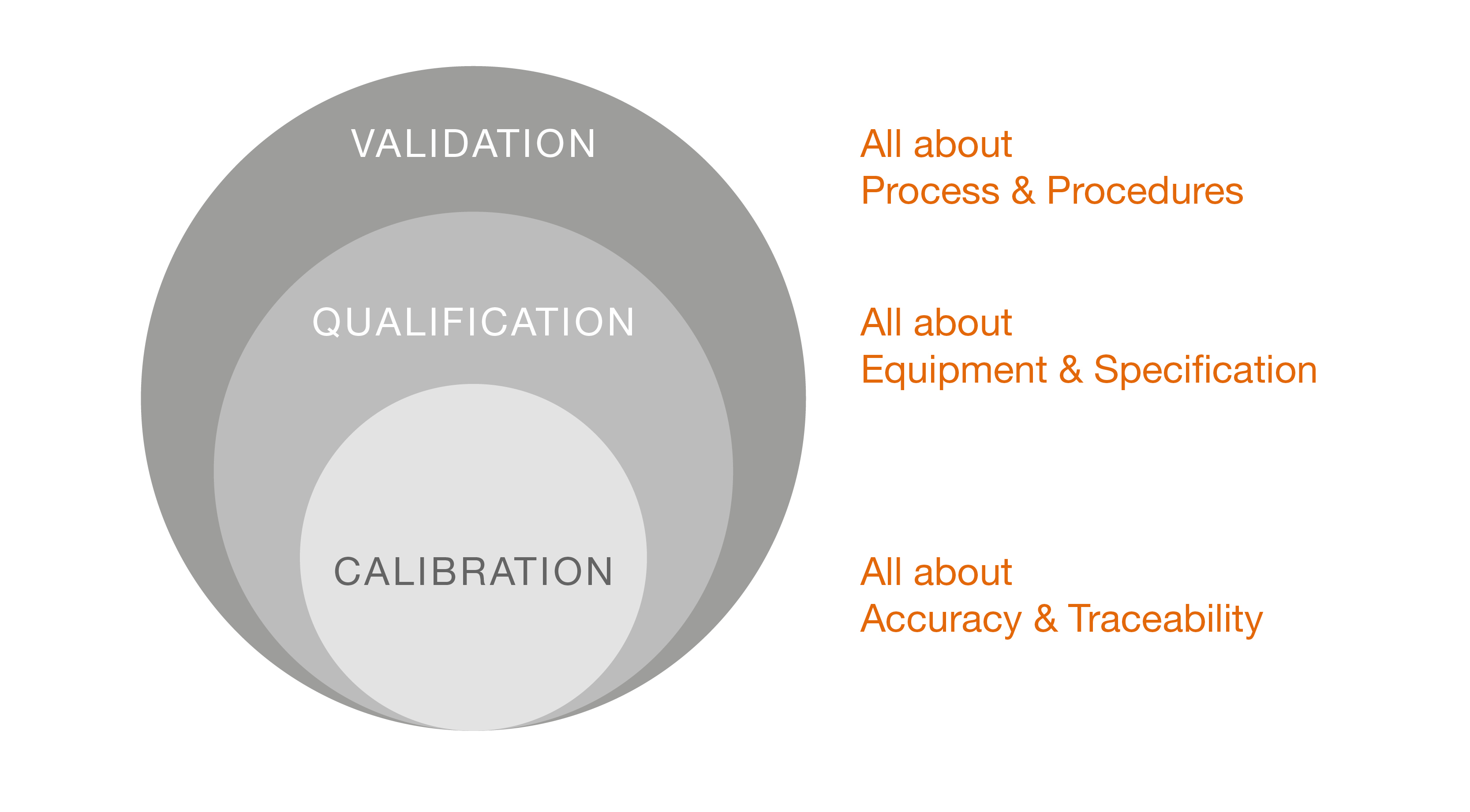
V
Validation Matrix
V
Validation Plan
V
Validation Protocol
V
Validation Report
V
VMP – Validation Master Plan
W
WFI – Water-for-Injection
W
WHO – World Health Organization
W
WIP – Washing-in-Place
Z
Z-Value
Z